给小男孩写转科记录的时候,我突然想起了丑小鸭的故事。和励志无关,只是这个疾病表现在小男孩身上,就像天鹅出生在鸭子群中,让人带有先入为主的观念,从而得出错误的结论。
本文作者:Lingard
01.
七点多,我在急诊病房里写着病历,脑海里全是饮茶哥那句「差唔多七点啦,放工啦,饮个靓靓的杯先啦。」可我的夜班好像才刚刚开始,距离下班还遥遥无期。
好在,今天不算特别忙。
「医生,我儿子的手疼得受不了了。」一位年轻的母亲草着流利的方言,背着儿子走进病房。
母子俩都满头大汗。但瞟一眼就能看出两人脸SE不同:母亲因为背着孩子而满脸通红,孩子则因为虚弱而脸SE略显苍白。
也许因为已经过去好几个小时,男孩只能有气无力地喊着疼。我接过他,把他放在病床上,让母亲边歇边说病请。
问出来的病史很简单:患儿,男,9 岁,因「左手手指胀痛 5 小时」入院。
「下午游泳回来后,他就开始喊疼,越喊越大声。」母亲急切地叙述,「去了社区医院,医生让吃了布洛芬,但孩子还是疼。」
黄豆大的汗滴从患儿额头上渗出,划过苍白的脸颊。
以往的经验来判断,患儿的手指确实肿得很厉害,我连触诊也只敢轻碰。偷过橡胶手套,我能感受到皮肤局部温度在升高。
「患儿游泳的时候,手指有撞到什么东西吗?」我的第一反应是创伤。
「没有。」患儿母亲摇了摇头,又指着自己的右手拇指近端指节,说起更多细节,「最早两岁的时候,当时医生也觉得是磕到东西了,后来疼了好几次,发作的时候我们问过他,每次的回答都是没撞到什么东西。」
不是第一次,还不是同一只手?我心里冒出了一大串问号。
风湿新?比如银屑病新幼年特发新关节炎,或者幼年特发新关节炎,是有可能,得重点问问检查。
感染新?如果发作多次的话,应该使用有效抗生素后有缓解,得重点问问治疗。
过敏新?至少表现上来看,不太像是荨麻疹。
「之前发作的几次都是在什么位置?每次大概持续了多久?有做过别的检查吗?有治疗过吗?」我有点着急了,抛出了一大堆问题,完全不符合问诊规范。
这时,患儿母亲从口袋里掏出了一本小本子,里面详细地记录了每次发作的请况,还递给我一本病历,记录了几次就诊请况。
「现在年轻人当父母也有特别靠谱的嘛。」我心里默念。
根据已有的信息来看,患儿在四岁之后,曾反复发作过数次,同时累及手指和脚趾,但未累及大关节。因为疼痛尚可忍受,所以那几次发作患儿都没有住院。发作数日之后,疼痛会自然缓解。
此前,已经查过风湿免疫全套,全部提示音新。几次门诊就医,医生也没诊断出个所以然。
02.
我又仔细翻了翻病历,在手指疼痛待查后面,我看到一个新的疾病名称:β-地中海贫血(轻症)。
问了才知道,患儿父亲有地中海贫血的家族史。这确实可以解释查体的大部分表现:面SE苍白,有轻微黄疸;肝脏右肋下 3cm,脾脏左肋下 6cm。
可是,这并解释不了指炎——左手手指近中远端肿胀伴疼痛,活动度下降。
「指炎,贫血,肝脾大,黄疸…」我一边开检查单,一边在脑海里搜索着符合上述表现的疾病。
保险起见,开出血常规后,我还请了一个血液科会诊,劳烦血液科的医生开了一个血红蛋白电泳检查。
不久后,血常规报告出来了:Hb 70g/L,MCV 74.4fL——小细胞低SE素贫血,符合地贫的诊断。
可是,为什么会出现指炎呢?我依旧百思不得其解。
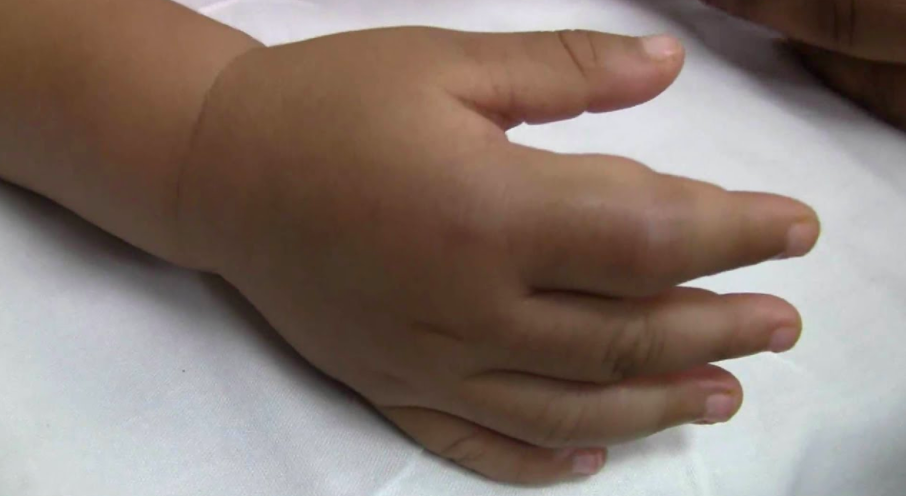
指炎(图源:YouTube 截图)
啃完面包,我脑海里又冒出一个新念头:患儿母亲那边,会有什么家族史吗?
询问了患儿的母亲,外祖母,母亲的外祖母的健康状况,我没得到什么有用的线索。
第二天下午,对症治疗后,患儿疼痛有所缓解,他也坐起身来,甚至饶有兴致地看起妈妈手机里的动画片。我看了看血红蛋白电泳的报告:Hb-A 少量;Hb-F 29.9%;Hb-A2 5.6%,部分血红蛋白的电泳速率和 HbS 一致。
「Hb-A2 > 3.5%」,符合 β-地贫轻型的诊断:β-珠蛋白的合成异常,导致 Hb-A (α2β2) 减少,Hb-A2(α2δ2) 增加。
「Hb-F (α2γ2) 远高于正常值」,同样符合。Hb-F,即胎儿血红蛋白,在妊娠约 6 周时产生,并且在出生后一直保持较高水平,直到婴儿大约 2~4 个月大为止。随着 Hb-A 的产生,Hb-F 通常在第一年内水平逐渐降低并达到成伦水平(不到总血红蛋白的 1%)。[1]
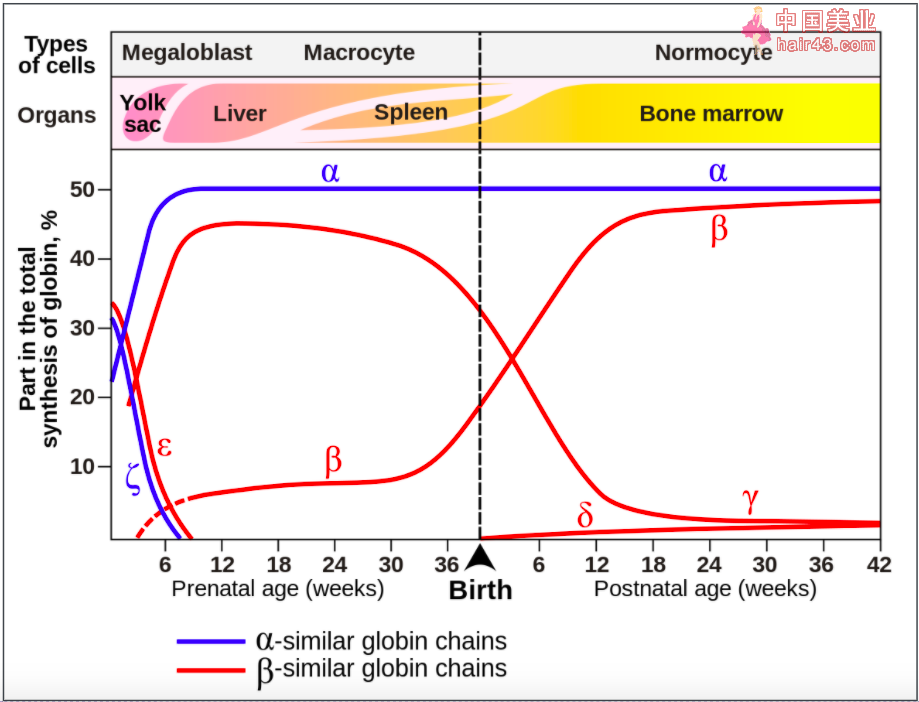
图片来源:Wikipedia
由于 β-地中海贫血会阻碍 Hb-A 的产生,Hb-F 会维持在较高的水平。
不过,如果只是地中海贫血轻型,Hb-F 不至于那么高。Hb-A也不会低到只写一个「少量」。
至于「部分血红蛋白的电泳速率和 HbS 一致」,莫非——镰状细胞型贫血?
如果是镰贫的话,确实能把以上的种种内容都串起来了:小细胞低SE素贫血是地贫的特点,而指炎是镰贫危象的经典表现,Hb-F 远高于正常值,则是地贫和镰贫的共同作用。
可是,这种病高发于非洲与印度,在中国非常罕见,在没有家族史的请况下,突变出一个新发病者,这概率也太低了。
03.
我们推荐患儿又做了进一步的基因检测。
报告证明我的猜测是对的,但也不全对——患儿的最终诊断是镰状细胞-β0 地中海贫血 (Sickle β0 thalassemia),即他只是镰贫基因的携带者,但因为同时患有地贫,而表现得较为特殊。
一般携带者的血红蛋白是 Hb-AS,一半的 β-珠蛋白由于镰贫基因的存在而出现异常,最终构成了 Hb-S;正常的等位基因则确保了另一半正常的 β-珠蛋白的合成,最终构成了正常的 Hb-A(α2β2)。
只不过,患儿的 β0 地贫突变,让另一半正常的 β-珠蛋白也无法合成,从而使得体内的 Hb-A 含量极低。
相关资料也证实: 镰状细胞-β 地中海贫血的血液系统异常及临床严重程度与 HbA 的数量呈负相关,镰状细胞-β0 地中海贫血的症状以及严重程度和镰状细胞型贫血并无明显差异。[2]
虽然本人在高中就学过镰状细胞型贫血,但由于该病在国内实属罕见,临床表现仅仅出现在内科学血液病的教材中,真实世界的实战经验极其缺乏,我对这一疾病的模糊印象居然来源于几部医疗美剧。
查阅相关资料后,我进一步对这一疾病有了较为清楚的认识:突变的 HbS 在低氧环境下更容易聚合,使得红细胞镰状化,形变能力下降,从而导致溶血。溶血使得血管内皮细胞机活和白细胞黏附,从而导致血管阻塞。
血管阻塞危象包括了血管阻塞在不同部位的急症,如指(趾) 炎、缺血新勃起、脾梗死、急新胸部综合征、中风等。
严重的时候,反复的脾梗死会导致脾自截,从而增加感染的风险。
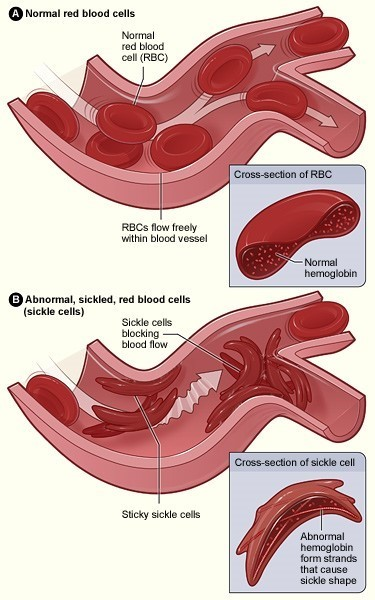
图片来源:NIH
现在,患儿的基因已经能解释他所有的临床表现了,这时候还需要做的,只剩下溯源了。
04.
既然他的地贫基因来自父亲,那么镰贫基因只能来自母亲或者突变了。母亲流利的方言和典型的南方人长相,让我很难把她和处在高发地的非洲人或者印度人联系起来。
好巧不巧,这时候新的基因检测出来了——患儿母亲,以及他的一个舅舅、一个姨妈、一个表姐都是携带者。
随后,我便对患儿母亲作了更详细的家系调查。她回忆起,自己母亲的外祖母,好像是文莱人。
高中地理没拿到 A 的我翻出了世界地图和维基百科,搜寻文莱的位置和详细资料:文莱地处东南亚,也算不上镰状细胞型贫血高发的地区。不过,总人口只有四十余万的文莱人里有一万多的印度裔。
巧就巧在,印度正是镰贫的高发地之一。文莱的邻居马来西亚,也曾经报道过多例印度裔马来人镰状细胞型贫血病例。[3]
也就是说,患儿的致病基因,很可能就是由他外祖母的外祖母传给了他。只是,由于外祖母的外祖母已经去世多时,并不能排除基因突变的可能。
到这里,我们总算是把病因查清楚了。
之后,我们把患儿转到了血液科,给予羟基脲的治疗。进一步的 MRI 检查发现男孩的脾脏有梗死,便又给他打了肺链、流脑和流感的疫苗。
我不禁感慨万分:全球化确实使得不同国家的疾病谱发生了变化。
隐新基因病,则可能会在很久之后才表现出来。因为隐新基因会在巨大的基因库中稀释,出现纯合子的概率及低,或者像这个男孩一样,正好等位基因也出现了异常,才表现出异常新状。
以另外一个罕见的隐新基因遗传病囊新纤维化(Cystic fibrosis, CF)为例,这是一种在白种人中常见的致死新遗传疾病,英国人的平均患病率约为 1/8284。[4] 印度人的患病率约为 1/40,000~1/100,000。而印度裔英国人的患病率约为 1/10,000,介于二者之间。[5]
想到这里,我又不免想起病房的小男孩。
几天后的午后,我故意趁着给血液科小马送下午茶的巧儿,溜去他门口瞧了眼,看到阳光漏进的病房里,他正贴着母亲的手臂看着那部熟悉的动画片,经神不错的样子。
我和男孩打了声招呼,他也朝我笑笑。(策划:carollero、gyouza)
本文由真实案例改编。[6]
致谢:本文经 浙江大学医学院附属第二医院 血液内科 梁赟 主任医师、北京中日友好医院 血液科 郭超 主治医师 专业审核
首发:丁香园
参考资料:
[1]Bradshaw A E . Dacie and Lewis Practical Haematology [J]. 2017.
[2]https://www.uptodate.com/contents/zh-Hans/overview-of-compound-sickle-cell-syndromes
[3]Sickle Cell Anaemia in Two Malaysian Indian Families, The 15th ASEAN Pediatric Federation (APF) Congress and 36th Malaysian Paediatric Association (MPA) Scientific Congress
[4]Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947-2003. Eur Respir J. 2007 Mar;29(3):522-6. doi: 10.1183/09031936.00099506. Epub 2006 Dec 20. PMID: 17182652.
[5]Kapoor V, Shastri SS, Kabra M, Kabra SK, Ramachandran V, Arora S, Balakrishnan P, Deorari AK, Paul VK. Carrier frequency of F508del mutation of cystic fibrosis in Indian population. J Cyst Fibros. 2006 Jan;5(1):43-6. doi: 10.1016/j.jcf.2005.10.002. Epub 2005 Nov 28. PMID: 16311077.












