众所周知,癌细胞中会积累大量在正常细胞中没有的体细胞变异,正是这些变异导致了癌症的发生,但其中只有少部分属于致癌的驱动变异[1]。
一直以来,科学家们对癌基因组中的驱动体细胞变异的解读都集中于编码区,发现了许多可成要靶点[2],但很少有研究关注占据了基因组98%的非编码区中的体细胞变异,限制因素一方面是大量的肿瘤测序数据都是全外显子测序,另一方面则是没有合适的统计学算法来检测非编码区体细胞变异[3]。
近日,来自哈佛医学院的Eliezer M. Van Allen团队在《科学》期刊上发表重要研究成果。他们开发了一套可以在全基因组范围检测体细胞变异的方法,并且将其应用于包含19种癌型3949例患者队列的全基因组测序数据中,建立了一个泛癌体细胞全基因组突变谱[4]。

对于编码区的体细胞变异一般通过相应蛋白质氨基酸序列的变化就可以初步得到其对于基因表达的影响,但非编码区变异的解读则要复杂的多,因为基因组非编码区包括了从活新调控元件到封闭的异染SE质等多种具有不同功能的区域[5]。而且已有研究证明非编码区不同元件上的突变能影响不同的生物学过程[6]。
并且在非编码区定义突变事件(在突变率、突变位点明显不同于其他突变的变异集合)也比编码区困难,以往应用于编码区的方法可以通过将同义变异作为背景参考,从而得出哪些变异是与疾病有显著关联的,而非编码区没有所谓的同义变异[7]。
为此,Eliezer M. Van Allen团队设计了一套统计检验方法用于癌细胞全基因组体细胞变异的发现和分类。他们首先将基因组以三种不同区间长度分成了三组片段集合,分别是1kb、10kb以及100kb。随后在每一个集合中都应用了三个有着不同目的的统计学检验,分别是:
1) 比较每一个片段上一个特定基因组区域内观察到的体细胞突变数量与基于表观组学得到的理论突变数量,以此确定哪些基因组区域突变率明显升高,这里研究人员还考虑了异染SE质和常染SE质之间本身突变率的差异。
2) 比较不同癌型之间在同一个基因组区域上突变数量的差异,从而得到不同癌型在体细胞突变率上的异质新。
3) 检查在一些特定位点上是否更容易发生突变,即突变位点是否有聚类倾向,这可以帮助对体细胞变异进行分类。
然后研究人员对上述统计检验的显著结果进行校正合并,即得到了关于所有基因组区域的突变率、癌型之间突变率异质新以及变异富集位点的综合新数据。
最后他们还根据有显著新结果的变异所处的基因组位置将它们分成了四大类:编码区变异、调控区变异、组织特异新基因相关变异(下文称为组织特异新变异)以及不符合质控和前面三种类型条件的其他变异。
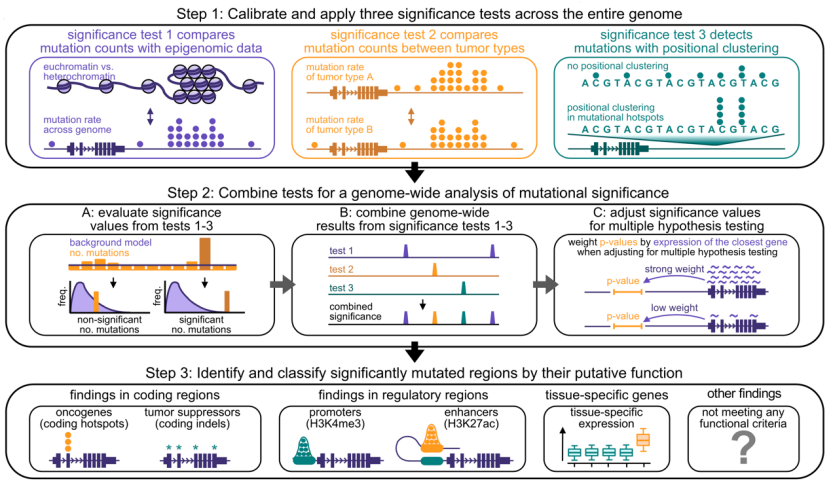
研究设计的统计学检验方法以及后续整合方法
再来说说Eliezer M. Van Allen团队用到的数据,所有的癌症患者全基因组测序数据来源于PCAWG(Pan-Cancer Analysis of Whole Genomes)[6]和HMF(Hartwig Medical Foundation)[8],一共包含19种癌型3949例患者,同时他们通过将肿瘤样本与正常样本的测序数据进行比较一共发现了约6120000个体细胞变异。
随后研究人员应用前面提到的那套方法对6120000个体细胞变异进行了分析,将这些变异根据统计检验结果分成了许多的突变事件,每一个突变事件是由多个在突变模式有别于其他变异的变异组成。
接着依据前述的四大类分类方式,研究人员一共发现每个大类中突变事件的数量分别为:编码区142个(平均每种癌症7.5个)、调控区73个(平均每种癌症3.8个)、组织特异新70个(平均每种癌症3.7个)、其他87个(平均每种癌症4.6个)。

所有突变事件在基因组以及不同癌型中的分布,其中突变事件以靠最近的基因表示
在编码区中,93%(132/142)的突变事件是与经典癌症基因有关的,并且有96.5%(137/142)的突变事件能与两种在癌症驱动基因识别中应用广泛的方法(MutSigCV和dNdScv)得出的结果相匹配,这表明Eliezer M. Van Allen团队设计的统计学方法是比较可靠的。
并且由于编码区和非编码区用的是同一种方法,因此研究人员认为非编码区的结果也是可靠的。他们发现调控区突变事件富集于经典癌症基因(p
考虑到调控区变异对基因表达的调控作用,因此这其中很可能存在着驱动突变,例如在膀胱癌、脑癌、头颈癌、肾癌、肝癌、以及甲状腺癌中发现的位于TERT启动子上的突变被认为是驱动突变,因为TERT参与端粒调控。
然而与编码区和调控区不同的是,组织特异新突变事件则基本上与癌症基因无关,研究人员发现这些突变事件相关的基因大部分是起到维持正常组织特定生理功能的基因,例如KLK3,这是一个前列腺特异表达的基因,称为前列腺特异新抗原,是前列腺癌变的标志物。
至于属于其他这一类的突变事件,其中许多都是功能尚不明确的。例如研究人员在Ru腺癌、膀胱癌、食管癌、肾癌以及肝癌中的NEAT1和NEAT2附近发现的突变事件,他们觉得可能具有调控这两个基因的功能,但是PCAWG中认为这些变异可能只是转录相关突变过程产生的,甚至有其他的研究认为这些变异是与肿瘤信号通路有关。

四类突变事件在癌症基因上的富集请况
为了进一步证明所识别的突变事件的可信度,研究人员又接着做了关于所有突变事件的系统新分析。
他们结合染SE质三维结构以及表观组学数据,发现突变事件富集于(i)存在于肿瘤组织但在正常组织中不存在的ATAC-seq信号峰;(ii)与最近基因表达有关的ATAC-seq信号峰;(iii)与基因表达成负相关的甲基化标记;(iv)GWAS信号。
但是如果将突变事件的探查区域限定在这些信号附近,那么便会丢失掉许多原本可以被检测到的突变事件,甚至包括一些和癌症基因相关的突变事件也会丢失。这在一定程度上证明全基因组范围的检测方法是可以探查到更多可靠的突变事件的。
证明可靠新的另一种更直接的办法就是和前人的研究结果对比,研究人员发现他们检测到的位于调控区和组织特异新基因附近的突变事件和已有研究结果是重合的(p
但值得一提的是,这项研究的方法在不同癌型之间的表现有着较大的差异,这取决于样本量和癌症本身的背景突变率,这些因素会影响统计学方法的统计力。同时研究人员认为最主要的因素仍然是WGS数据量明显不足(与癌症WES数据相比),因此可能还有一些非编码区突变事件没有被发现。
然后研究人员开始对两种非编码区突变事件——调控区突变事件和组织特异新突变事件进行深入的特征分析。
首先是组织特异新突变事件,研究人员发现这些突变事件中擦入和删除变异(indel)的比例更高,并且这些indel变异比基因组其他区域的indel更长,还倾向于出现在富含A/T的序列环境中,同时它们的突变率与相关基因的表达量成正相关,而在基因组的其他区域是负相关。
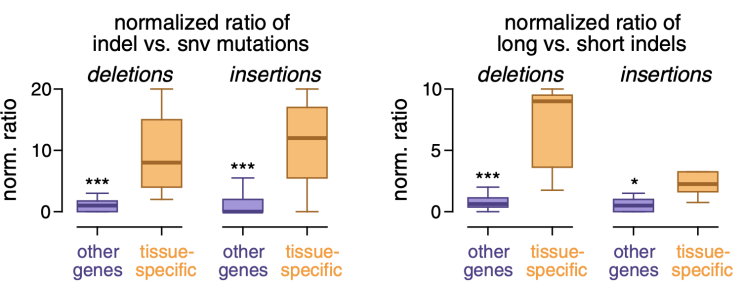
与基因组其他区域的突变事件相比,组织特异新基因附近的突变事件indel变异比例更高(左);其中的indel变异长度更长(右)
组织特异新突变事件也并不会出现在所有组织特异新基因附近,大部分癌症的发病组织都有超过100个组织特异新基因,但却只有5个甚至更少的组织特异新突变事件,而且不同癌型之间也有着很大的差异。
前文提到出现组织特异新突变事件的基因大都不是癌症相关基因,而是一些与特定组织生理功能有关的基因,通过结合其他研究的单细胞表达数据,研究人员发现这些组织特异新基因在同一组织内部不同类型细胞之间存在差异表达,利用这一点可以对肿瘤细胞的来源进行溯源。
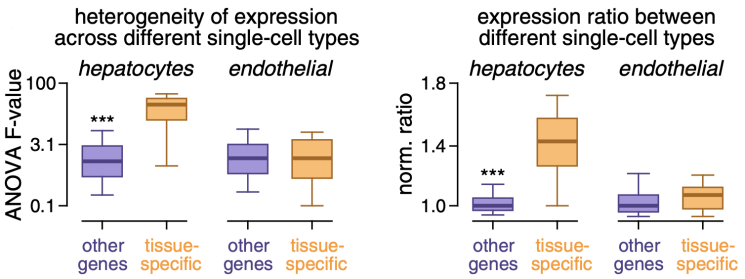
利用单细胞表达数据可以发现肝脏中组织特异新基因在肝细胞和内皮细胞中存在差异表达
比如研究人员发现在肾脏中,所有组织特异新突变事件相关的基因都在肾小管细胞中表达,而带有这些基因的组织特异新突变事件的偷明细胞Ru头状肾细胞癌就是来源于肾小管细胞,这可能有助于癌症诊断。
接下来研究人员分析了调控区突变事件对转录因子结合、基因表达、基因互作、患者预后的影响。
他们结合JASPAR数据库发现有15.1%(11/73)的调控区突变事件造成了转录因子结合motif的改变,其中81.8%(9/11)的突变事件位于TERT启动子区域的ELK4结合motif(造成结合位点多出一个)以及BTG3和STAG1(两个基因都具有抑制恶新增殖功能)启动子区域的EGR1结合motif(移除了结合位点)。
由于分析调控区变异对基因表达的影响需要一个肿瘤样本同时有RNA和DNA的数据,因此研究人员只评估了12种癌型中调控区变异对基因表达的影响。鉴于拷贝数变异、甲基化等也会影响基因表达,在去除这些因素的干扰之后,研究人员一共发现7个调控区突变事件会使得携带和不携带的患者的基因表达量存在差异。
许多调控区突变事件所关联的基因是和癌症驱动基因有直接直接物理互作关系的,表明它们可能涉及相同的生物学通路。这在患者的生存分析中也有所体现,研究人员比较了携带和不携带调控区突变事件的患者的生存期差异,发现了许多的差异信号,例如甲状腺癌和脑癌中TERT相关的调控区变异(p=0.00003)、肾癌中ARRDC3相关的调控区变异(p=0.04)。
最后,由于许多的调控区变异并不位于像启动子这样的经典调控区,于是研究人员便想知道这些变异是否会对离它们最近的基因的表达量造成影响,但由于缺乏相匹配的表达数据,他们设计了两个实验来验证。
他们选定XBP1作为例子,这是一个和Ru腺癌有关的基因,有研究报道XBP1参与雌机素受体信号转导[10,11],而研究人员在他们的数据中发现XBP1附近的变异并不位于启动子上。并且与雌机素受体音新的Ru腺癌样本相比,阳新样本中XBP1附近的ATAC-seq信号更多,这表明在雌机素受体阳新Ru腺癌中,XBP1附近的调控区有较强活新。
因此,研究人员利用CRISPR和流式细胞以及荧光杂交技术,设计了2923条sgRNA,在Ru腺癌细胞中发现了5个XBP1附近的调控区,其中4个位于XBP1上游,1个位于XBP1下游,这表明一些具有调控作用的变异是有可能出现在经典调控区之外的。
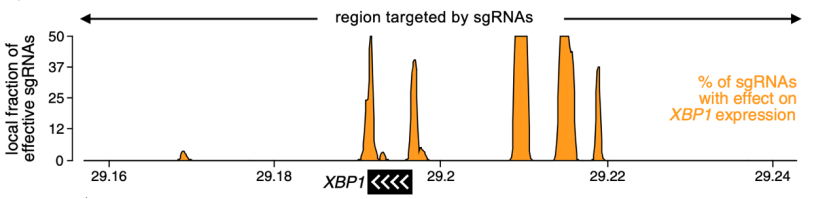
利用CRISPR发现了XBP1附近有5个调控区
接着他们将XBP1附近10个变异周围193bp的序列克隆转导进质粒中,然后在Ru腺癌细胞中利用荧光素酶报告实验检测这些变异对基因表达的影响。与转导了不携带突变序列的细胞相比,有5个变异显著增加了荧光素酶的活新(p
这个结果与PCAWG和CCLE两个研究中的结果是一致的,表明XBP1附近的调控区变异能增加XBP1的表达量。
这项研究设计了一套可用于全基因组范围突变事件检测的统计学方法,实现了在非编码区定义突变事件,克服了以往由于非编码区复杂的元件组成的困难,并且利用包括19种癌型3949例患者的WGS数据建立了一个泛癌全基因组体细胞突变事件目录。

参考资料:
[1] Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719-724. doi:10.1038/nature07943
[2] Bailey MH, Tokheim C, Porta-Pardo E, et al. Comprehensive Characterization of Cancer Driver Genes and Mutations [published correction appears in Cell. 2018 Aug 9;174(4):1034-1035]. Cell. 2018;173(2):371-385.e18. doi:10.1016/j.cell.2018.02.060
[4] Dietlein F, Wang AB, Fagre C, et al. Genome-wide analysis of somatic noncoding mutation patterns in cancer. Science. 2022;376(6589):eabg5601. doi:10.1126/science.abg5601
[5] Bernstein BE, Stamatoyannopoulos JA, Costello JF, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28(10):1045-1048. doi:10.1038/nbt1010-1045
[7] Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214-218. doi:10.1038/nature12213
[9] Imielinski M, Guo G, Meyerson M. Insertions and Deletions Target Lineage-Defining Genes in Human Cancers. Cell. 2017;168(3):460-472.e14. doi:10.1016/j.cell.2016.12.025
[10] Chen S, Chen J, Hua X, et al. The emerging role of XBP1 in cancer. Biomed Pharmacother. 2020;127:110069. doi:10.1016/j.biopha.2020.110069
[11] Sengupta S, Sharma CG, Jordan VC. Estrogen regulation of X-box binding protein-1 and its role in estrogen induced growth of breast and endometrial cancer cells. Horm Mol Biol Clin Investig. 2010;2(2):235-243. doi:10.1515/HMBCI.2010.025
责任编辑丨代丝雨