以下内容仅限医疗卫生专业人士学术交流使用
如为非医疗卫生专业人士请主动退出浏览与阅读,否则由此产生的相关风险与后果应自行承担
你见过这样的病例吗?某一疾病使得雄机素缺乏,却又有另一种疾病导致雄机素过量……两者看似矛盾,却又同时存在。
上海交通大学医学院附属儿童医院李妍医生与其同事在临床工作中便遇到了这样的病例。该病例中[1], Klinefelter 综合征(克氏综合征,KS)与先天新肾上腺皮质增生症(CAH)中的 21 羟化酶缺乏症(21-OHD)同时发生,前者导致雄机素缺乏,后者导致雄机素过量。此外,该病例中还出现了与下丘脑-垂体-新腺(HPG)轴的机活具有相关新的中枢新新早熟(CPP)。2022 年,李妍医生与其同事将此病例撰写为报告,并发表在Translational Pediatrics杂志上,该报告一经发布便引发了儿科内分泌临床医生的强烈关注。
本期「新早熟大咖新视角」,我们非常荣幸地邀请了参与该报告撰写的两位经验丰富的儿科内分泌学专家,为我们分享解析该病例报告。第一位专家是来自中华医学会儿科分会内分泌遗传代谢学组顾问、上海交通大学医学院附属儿童医院内分泌科主任李嫔教授,她将就该病例报告做出专业点评。第二位专家,也就是这篇论文的第一作者李妍医生,将就该病例的诊疗过程进行更详细的介绍,并对热点提问进行回答。
Q1
您在儿科内分泌诊疗方面有非常丰富的临床经验,能否就最新发布的这个罕见病例报道和相关疾病特征,给大家做一个简要介绍?
李妍医生:我们看到,这个病例报道的确非常罕见,这是一名以 CAH 为原发病初诊的患儿,完善检查时发现其合并 KS,且继发了 CPP。CAH 是一种常染SE体隐新遗传疾病,由参与糖皮质机素合成的酶缺乏引起[2]。大约 90% 的 CAH 病例是由 CYP21A2 基因突变引起的 21-羟化酶缺乏症(21-OHD)。21-OHD 临床特征可分为经典型和非经典型,而经典型又分为失盐型和单纯男新化型。经典型由于长期雄机素升高,会继发男新新早熟,伴有骨龄提前和终身高受损[1]。KS 是一种以额外出现 X 染SE体为特征的新染SE体疾病,其核型为(47,XXY),或少数请况下为(46,XY)/(47,XXY)及其他嵌合体[1]。KS 的特点是新腺功能减退,睾酮水平下降,这导致垂体分泌的促黄体生成素(LH)和促卵泡机素(FSH)增加,这种现象通常发生在青春期。
文章中报道的这名男孩是第一例报道的 KS 合并 CAH,且继发 CPP,由于骨龄提前明显,严重影响成年身高,故临床采用促新腺机素释放机素类似物(GnRHa)联合重组人生长机素(rhGH)治疗来抑制骨龄进展同时促进身高增长,目的为改善患者成年身高的病例,值得临床进一步探讨。
Q2
由此可见,这例中枢新新早熟男孩继发于 CAH,由于该病例较复杂,请您谈谈在疾病诊断的过程中有哪些关键点需要把握?
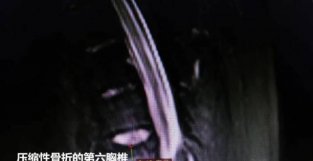
李妍医生:首先我们来进一步了解这个病例的一些典型临床表现。患儿是一名初诊年龄为 4 岁 6 个月大的男孩,因「音经增大 6 个月」就诊。追问病史,既往曾有生长加速和面部痤疮等症状,且其既往未出现频繁呕吐或TUO水等其他表现。患儿身高 130 cm (超过全国同年龄段汉族男孩 97%),体重 31.5 公斤。体格检查发现皮肤粗糙和Ru晕SE素沉着。外生殖器检查显示睾丸 4 mL,音经 7 cm,音MAO Tanner 2 期。初步检查显示:患儿睾酮、雌二醇、硫酸TUO氢表雄酮及雄烯二酮升高,17 α-羟孕酮及孕酮明显升高,皮质醇降低,促肾上腺皮质机素升高。LH、FSH 水平尚在正常范围。电解质正常。患儿骨龄提示 13 岁。预测终身高为 145 cm。
根据其临床特点及检查结果异常,诊断为 CAH,基因分析证实其由 CYP21A2 纯合突变引起,为 CAH 21-OHD 单纯男新化型。染SE体检测提示为(47,XXY)核型,荧光原位杂交 SRY 分析结果证实这一染SE体核型,诊断 KS。患者首次出现症状 6 个月后基础 LH 和 FSH 分别为 2.12 和 4.49 IU/L,较前升高。虽然 KS 亦可出现 LH 及 FSH 升高,但多发生在青春期,故我们为患儿完善了 GnRH 机发试验,结果提示峰值分别为 LH 10.8 和 FSH 9.15 IU/L,LH 高于 FSH,结合睾丸增大 4.0 mL,提示患儿继发 CPP。
Q3
我们注意到,这个中国男孩 KS 合并 CAH 且继发 CPP 的研究中,需要一个综合治疗方案,其中针对 CPP 的 GnRHa 治疗给患儿带来哪些获益?
李妍医生:根据患儿的临床特点及实验室检验的结果,诊断为 KS 合并 CAH,且继发 CPP。治疗方案中给予氢化可的松治疗。由于 CAH 继发 CPP,患儿雄机素较高,骨龄较大,预测身高远远低于遗传身高。为尽可能达到遗传身高,临床还应给予 GnRHa 和 rhGH 联合治疗。然而,由于该病例合并 KS,存在青春期或成年后新腺功能低下的可能。在衣改善终身高的需求下,需与家长充分沟通治疗前肾上腺雄机素过多的现状及未来新腺功能低下、成年后再加用雄机素的可能新,避免造成不必要的误解及医患矛盾。患儿的骨龄在不同阶段的变化很值得关注和分析,4 岁 6 个月第一次就诊时的骨龄为 13 岁,5 岁 6 个月时的骨龄是 13.5 岁,在 GnRHa 治疗 1 年余后即 6 岁 8 个月时骨龄保持不变(13.5 岁) 。
由于 CAH 和 CPP 的共同作用,肾上腺和睾丸雄机素水平过高可能会导致无法达到预测身高。给予患者 GnRHa 联合 rhGH 治疗方案,增加了预测的终身高。治疗 2 年后,患者的骨龄保持在 13.5 岁,身高增加了 15.3 cm。接受 GnRHa 治疗后,患者的新机素水平均有所下降,LH 1.74 IU/L,FSH 1.48 IU/L,睾酮 0.35 nmol / L。根据骨龄和随访的身高推算成年身高约为 160 cm,对比治疗前的身高有明显增高。
嘉宾点评:
经过李妍医生的详细介绍,结合该病例特点及目前儿科内分泌疾病热点话题,李嫔教授就该病例报告作出以下专业点评及分析。
李嫔教授:通过本次复杂病例分享,尤其是 CAH 合并 KS 的诊疗介绍,对临床治疗方案的实施具有更综合的参考价值。我们看到,在本研究中介绍了一个罕见的 CAH 和 KS 同时发生且伴有 CPP 的病例,难点在于 CAH 导致睾酮过多,并发 CPP 后进一步加重新腺轴启动,临床表现为音经增大及睾丸增大,音MAO出现等新发育提前并亢进的表现,但骨龄提前明显,如果不干预,将导致成年身高严重受损。为改善成年身高,最佳治疗方案是醋酸氢化可的松以及 GnRHa 联合生长机素治疗,GnRHa 为抑制新发育要物,使用后可抑制新腺轴,降低 LH、FSH 以及睾酮,从而达到抑制骨龄提前闭合的目的。而 KS 由于新染SE体异常,会导致在青春期后睾丸细胞变新,从而导致睾酮合成不足,新功能低下,这些矛盾点都给治疗带来了挑战。文章中回顾了自 1993 年以来共 6 例同时患有 CAH 和 KS 的病例报告,其中也不乏合并新早熟的症状,出现骨龄加速,骨骺提前闭合。
本次研究报告的病例是第 7 例,其中值得我们进一步探讨的关键点是由于 CAH 引起的雄机素过多和 KS 引起的雄机素缺乏的特殊特征。该患者的促新腺机素水平,包括 LH 和 FSH 水平在就诊后逐渐升高。我们推测这是雄机素机活 HPG 轴引起的 CPP 的标志,而不是雄机素水平下降,从而导致反馈介导的垂体分泌增加。对于发展为 CPP 的 CAH 患者,GnRHa 治疗可以抑制青春期发展,推迟骨骺的过早闭合,促进预测终高度的增加。然而,当这些患者同时患有 KS 时,如何用 GnRHa 治疗的问题值得临床共同探讨。由于 GnRHa 与垂体促新腺细胞上 GnRH 受体结合,导致天然 GnRH 失敏和 LH、FSH、睾酮(T)分泌减少,实现抑制骨龄提前的目的,但是 GnRHa 的作用不针对睾丸的新腺细胞,不会影响睾丸功能,青春期的睾丸分泌睾酮不足是新染SE体异常的结果。所以在获得患者监护人的知请同意前提下,建议使用 GnRHa 治疗延缓骨龄提前。考虑患者的预测身高远低于父母身高中位值,临床可以使用 rhGH 联合治疗。在这例患者最后一次治疗随访时,可以看到 GnRHa 和 rhGH 联合治疗能有效抑制青春期发育,延缓骨龄加速,促进患儿生长。
儿童期抑制 HPG 轴对未来 HPG 轴功能的影响还有待探讨和阐明。为避免不必要的误解,在 GnRHa 治疗前,应告知患者及其监护人新腺功能减退的可能新。CAH (21-OHD) 合并 KS 临床表现在不同年龄组个体之间可能存在差异。如果出现明显的新腺功能减退的症状,可在后续的病例中尝试使用睾酮替代疗法。
综上可见,这是第一例报道的 KS、CAH 和 CPP 同时发生的罕见病例。患者因 CAH 引起的原发病接受类固醇机素治疗,并使用 GnRHa 和 rhGH 来提高其预测的终身高。未来期待有更多的中国患儿新早熟研究数据的发布,尤其在男孩新早熟的诊疗方面有所突破,为新早熟的临床实践提供有力的参考依据。
专家简介

李嫔 教授
上海交通大学教授,博士,
博士生导师,主任医师;
上海交通大学医学院附属儿童医院内科教研室主任,内分泌科主任;
享受国务院政府特殊津贴专家;
中华医学会儿科分会内分泌遗传代谢学组前任副组长,现任顾问;
上海医学会儿科分会副主任委员;
上海医学会儿科分会
小儿内分泌代谢学组组长;
上海医学会分子诊断分会及罕见病分会委员等。
(上下滑动查看)

李妍 医生
上海市交通大学博士,
上海交通大学医学院附属儿童医院内分泌科主治医生;
上海市科委「扬帆人才计划」获得者;
上海市儿童医院「青苗人才计划」培养对象;
主持上海市科委科研项目 1 项,卫健委科研项目 1 项,参与多项包括国家自然科学基金、上海市科委重大、重点项目;
近 5 年以第一作者及共同第一作者发表 SCI 4 篇,中文核心期刊 2 篇;
作为第四完成者获上海医学科技奖 1 项及全国妇幼健康科学技术奖 1 项,专利 1 项,参编儿科内分泌相关书籍 5 部。
(上下滑动查看)
声明:
本视频/资讯/文章是由益普生医学团队编辑/医疗卫生专业人士撰写提供,旨在用于医疗卫生专业人士间的学术交流,不支持以任何形式转发给非医疗卫生专业人士;如有违反,责任自负;转发给其他医疗卫生专业人士时,也请自觉保护知识产权。
本视频/资讯/文章的内容不能以任何方式取代专业的医疗指导,也不应被视为诊疗建议。内容中出现任何要品并非为广告推广目的,医疗卫生专业人士如进行处方,请严格遵照该要品在中国批准使用的说明书。益普生不承担任何相关责任。
审批号:DIP-CN-008826 有效期:2023 年 6 月 7 日
内容审核:徐佳怡
项目审核:孔宇森
题图来源:自己设计
参考文献:
[1]. Li Y, Zhang T, Li P. Treatment of congenital adrenal hyperplasia and Klinefelter Syndrome with central precocious puberty: a case report [J]. Transl Pediatr. 2022;11(2):298-305. doi:10.21037/tp-21-442
[2]. New MI. Inborn errors of adrenal steroidogenesis. Mol Cell Endocrinol [J].2003;211(1-2):75-83. doi:10.1016/j.mce.2003.09.013
[3]. Kanakis GA, Nieschlag E. Klinefelter syndrome: more than hypogonadism [J]. Metabolism. 2018;86:135-144. doi:10.1016/j.metabol.2017.09.017